กลุ่มอาการพราเดอร์-วิลลี่ (Prader-Willi Syndrome)
1. เด็กกลุ่มอาการพราเดอร์-วิลลี่มักมีปัญหาเกี่ยวกับการรับประทานอาหารได้น้อย ความอยากอาหารน้อย ความตึงตัวของกล้ามเนื้อต่ำมาก น้ำหนักตัวน้อยในช่วงประมาณ 2 ขวบปีแรก จากนั้นจะเริ่มรับประทานอาหารมากขึ้นและรู้สึกอิ่มน้อยลงเรื่อย ๆ นำไปสู่โรคอ้วนและภาวะแทรกซ้อน เช่น เบาหวาน ไขมันในเลือดสูง ในเวลาต่อมา หากไม่ได้รับการดูแลเรื่องโภชนาการอย่างเหมาะสม
2. มีลักษณะใบหน้าที่จำเพาะ และมักมีความบกพร่องทางด้านสติปัญญาร่วมกับปัญหาพฤติกรรมและอารมณ์ 3. มักพบความผิดปกติของระบบต่อมไร้ท่อ เช่น ขาดฮอร์โมนการเจริญเติบโต ไทรอยด์ฮอร์โมนต่ำ กลุ่มอาการพราเดอร์-วิลลี่คืออะไร กลุ่มอาการพราเดอร์-วิลลี่เกิดจากความผิดปกติทางพันธุกรรม โดยทำให้เกิดความผิดปกติของสมอง โดยเฉพาะส่วนที่ทำหน้าที่ควบคุมการผลิตฮอร์โมนต่าง ๆ เช่น ไทรอยฮอร์โมน ฮอร์โมนการเจริญเติบโต อาการของโรคมีความแตกต่างไปตามช่วงอายุ ได้แก่ 1. ช่วงอายุ 0-9 เดือน มีความตึงตัวของกล้ามเนื้อต่ำ ดูดนมได้น้อย เนื่องจากไม่มีแรงดูด มักต้องใส่สายให้อาหารทางจมูก 2. ช่วงอายุ 9 - 25 เดือน พัฒนาการล่าช้า เริ่มรับประทานอาหารได้มากขึ้น การเจริญเติบโตเริ่มอยู่ในเกณฑ์ปกติ 3. ช่วงอายุ 2 - 4 ปี พัฒนาการล่าช้า รับประทานอาหารได้ปกติ น้ำหนักมีแนวโน้มเพิ่มมากขึ้น 4. ช่วงอายุ 4 - 8 ปี พัฒนาการล่าช้าหรือบกพร่องทางสติปัญญา มีพฤติกรรมการรับประทานอาหารมากผิดปกติ รู้สึกอิ่มยากมากขึ้น แต่ยังรู้สึกอิ่มได้ หากไม่มีการปรับอาหารหรือปรับพฤติกรรมการรับประทานอาหารมักมีปัญหาอ้วนตามมา 5. ช่วงอายุ 8 - 13 ปี มีภาวะบกพร่องทางสติปัญญา ส่วนมากมักมีสติปัญญาอยู่ในระดับบกพร่องเล็กน้อย (IQ เฉลี่ย 60 - 70) ส่วนน้อยมีสติปัญญาคาบเส้น (borderline IQ) หรือต่ำกว่าเกณฑ์เฉลี่ย (IQ 80-89) บางรายมีปัญหาพูดไม่ชัด มีพฤติกรรมการรับประทานอาหารมากผิดปกติ รู้สึกอิ่มยากมากขึ้น 6. ช่วงอายุ 13 ปี จนถึงวัยผู้ใหญ่ มีลักษณะการเข้าสู่วัยรุ่นและลักษณะทางเพศของร่างกายผิดปกติ จากฮอร์โมนเพศต่ำ มีภาวะบกพร่องทางสติปัญญา มีพฤติกรรมการรับประทานอาหารมากผิดปกติ รู้สึกอิ่มยากมากหรือไม่รู้สึกอิ่ม มีภาวะอ้วนหากไม่ได้ปรับพฤติกรรมการรับประทานอาหารอย่างเหมาะสม มีปัญหาพฤติกรรมต่าง ๆ เช่น ชอบแกะเกาผิวหนัง ย้ำคิดย้ำทำ วิตกกังวล เป็นต้น
เกิดจากความผิดปกติเกี่ยวกับการแสดงออกของยีนบริเวณแขนยาวของโครโมโซมคู่ที่ 15 ซึ่งปกติจะมีการแสดงออกเฉพาะโครโมโซมที่ได้รับมาจากบิดา โดยความผิดปกติที่ทำให้ยีนบริเวณดังกล่าวไม่แสดงออก ได้แก่ 1. มีการขาดหายไปของชิ้นส่วนแขนยาวของโครโมโซมคู่ที่ 15 บริเวณ 15q11.2-q13 (พบได้บ่อยที่สุด ประมาณร้อยละ 70 ของผู้ป่วย) 2. ได้รับชิ้นส่วนแขนยาวของโครโมโซมคู่ที่ 15 ทั้ง 2 ข้างมาจากมารดา ซึ่งไม่มีการแสดงออกของยีนส์บริเวณดังกล่าว 3. ความผิดปกติของบริเวณที่ควบคุมการแสดงออกของยีนบนแขนยาวของโครโมโซมคู่ที่ 15 ทำให้ไม่มีการแสดงออกของยีนส์บริเวณดังกล่าว
ทำได้โดยการตรวจหาความผิดปกติของพันธุกรรม โดยมีเทคนิคการตรวจหลายวิธี เช่น 1. การตรวจดูการแสดงออกของพันธุกรรมบริเวณแขนยาวของโครโมโซมคู่ที่ 15 (Methylation-specific PCR) ซึ่งผู้ป่วยจะตรวจไม่พบการแสดงออกของพันธุกรรมบริเวณ 15q11.2-q13 ที่ได้รับมาจากบิดา 2. การตรวจหาการขาดหายไปของพันธุกรรมบริเวณแขนยาวของโครโมโซมคู่ที่ 15 โดยใช้สารเรืองแสง (Fluorescence in situ hybridization analysis of 15q11.2-q13) 3. SNP microarray สามารถตรวจหาการขาดหายไปของพันธุกรรมบริเวณ 15q11.2-q13 และตรวจหาความผิดปกติกรณีผู้ป่วยได้รับโครโมโซมคู่ที่ 15 มาจากมารดาทั้ง 2 ข้าง (maternal uniparental disomy) 4. MS-MLPA สามารถตรวจหาการขาดหายไปของพันธุกรรมบริเวณ 15q11.2-q13 และบริเวณที่ควบคุมการแสดงออกของพันธุกรรมบริเวณ 15q11.2-q13 (imprinting center and SNORD116 deletion)
ปัจจุบันยังไม่มีวิธีรักษาความผิดปกติพันธุกรรมที่พบในโรคนี้ให้หายขาด การรักษาจึงเน้นการดูแล รักษา ฟื้นฟูความผิดปกติที่พบ ได้แก่
1.อาการรับประทานอาหารได้น้อยในช่วงทารกและเด็กเล็ก พิจารณาใช้อุปกรณ์ช่วย เช่น จุกนมชนิดพิเศษ (Nuk nipple หรือ Haberman nipple) หรือการใส่สายให้อาหารทางจมูก (nasogastric tube) โดยพิจารณาถอดสายออกเมื่ออายุมากขึ้นและสามารถรับประทานอาหารได้เพียงพอสำหรับการเจริญเติบโต ส่วนมากสามารถถอดออกได้เมื่ออายุไม่เกิน 6 เดือน (บางรายจำเป็นต้องใส่สายให้อาหารจนถึงอายุ 1 ปี) 2. ให้การส่งเสริมและกระตุ้นพัฒนาการในเด็กที่มีพัฒนาการช้า โดยแนะนำให้เริ่มฝึกกระตุ้นพัฒนาการให้เร็วที่สุดเท่าที่เป็นไปได้ 3. ภาวะฮอร์โมนเพศต่ำ พิจารณาให้การรักษาโดยให้ฮอร์โมนเสริมโดยแพทย์ผู้เชี่ยวชาญด้านต่อมไร้ท่อ ส่วนภาวะอัณฑะไม่ลงถุงในผู้ป่วยเพศชาย พิจารณาผ่าตัดรักษากรณีไม่ตอบสนองต่อการรักษาด้วยฮอร์โมน โดยแนะนำให้ผ่าตัดรักษาภายในช่วงอายุ 6-18 เดือน 4. ภาวะขาดฮอร์โมนการเจริญเติบโต พิจารณาให้การรักษาโดยการให้ growth hormone โดยแพทย์ผู้เชี่ยวชาญด้านต่อมไร้ท่อ 5. ดูแลพฤติกรรมการรับประทานอาหาร หลังจากผู้ป่วยเริ่มรับประทานอาหารได้มากขึ้นโดยนักกำหนดอาหารหรือแพทย์ผู้เชี่ยวชาญด้านโภชนาการ เพื่อปรับอาหารให้ตรงกับความต้องการพลังงานในแต่ละวัย ป้องกันภาวะอ้วนในอนาคต 6. ติดตามและประเมินพฤติกรรมผิดปกติที่อาจพบ เช่น พฤติกรรมชอบแกะเกาผิวหนัง ปัญหาทางอารมณ์ โดยกุมารแพทย์ผู้เชี่ยวชาญด้านพัฒนาการและพฤติกรรมหรือจิตแพทย์เด็ก 7. ตรวจประเมินสายตาเป็นระยะ เพื่อเฝ้าระวังปัญหาเกี่ยวกับการมองเห็น
สำหรับการดูแลติดตามต่อเนื่องในกลุ่มอาการพราเดอร์-วิลลี มีรายละเอียดดังนี้
1. กลุ่มอาการพราเดอร์-วิลลี มีความผิดปกติหลายระบบ จึงจำเป็นต้องได้รับการติดตามและดูแลโดยทีมสหสาขาวิชาชีพ เพื่อดูแลรักษาและป้องกันความผิดปกติต่าง ๆ ที่อาจตามมา
2. โดยทั่วไปเป็นความผิดปกติที่เกิดขึ้นเองโดยบังเอิญ โอกาสเกิดซ้ำในบุตรคนถัดไปน้อยกว่าร้อยละ 1 3. เนื่องจากผู้ป่วยมากกว่าร้อยละ 90 มีพัฒนาการช้า จึงจำเป็นต้องได้รับการประเมินและกระตุ้นพัฒนาการ โดยควรเริ่มให้เร็วที่สุด โดยเฉพาะอย่างยิ่งภายใน 3 ขวบปีแรก ซึ่งจะทำให้แนวโน้มพัฒนาการดีกว่า ได้รับการส่งเสริมพัฒนาการหลังอายุ 3 ปี 4. ช่วงวัยเด็กเล็กผู้ป่วยมักรับประทานอาหารได้น้อยมาก แต่เมื่ออายุมากขึ้นจะรับประทานอาหารได้มากขึ้นเรื่อย ๆ จนสุดท้ายมีภาวะรับประทานอาหารมากผิดปกติ ดังนั้นผู้ดูแลจึงควรดูแลเรื่องอาหารที่ผู้ป่วยรับประทาน ให้รับประทานแต่อาหารที่มีประโยชน์ ไม่รับประทานขนมขบเคี้ยวหรือขนมหวาน เนื่องจากจะทำให้ได้รับพลังงานมากเกินไปและทำให้อ้วน 5. ปัจจุบันมีชมรมพราเดอร์-วิลลี่ประเทศไทย ซึ่งเป็นการรวมตัวกันของกลุ่มผู้ปกครองของผู้ป่วยพราเดอร์-วิลลี่ เพื่อให้ข้อมูล ความช่วยเหลือ และแบ่งปันประสบการณ์ในการดูแลผู้ป่วย โดยผู้ที่สนใจสามารถติดต่อได้ทางช่องทางออนไลน์ต่าง ๆ เช่น facebook page 6. เนื่องจากการเจริญเติบโตของผู้ป่วยพราเดอร์-วิลลี แตกต่างจากเด็กทั่วไป จึงจำเป็นต้องได้รับการติดตามโดยใช้กราฟการเจริญเติบโตสำหรับกลุ่มอาการพราเดอร์-วิลลี โดยเส้นประแทนค่าเฉลี่ยการเจริญเติบโตของเด็กทั่วไป ดังนี้
กราฟน้ำหนักตามอายุของเด็กกลุ่มอาการพราเดอร์-วิลลี เพศชาย (A) และเพศหญิง (B) 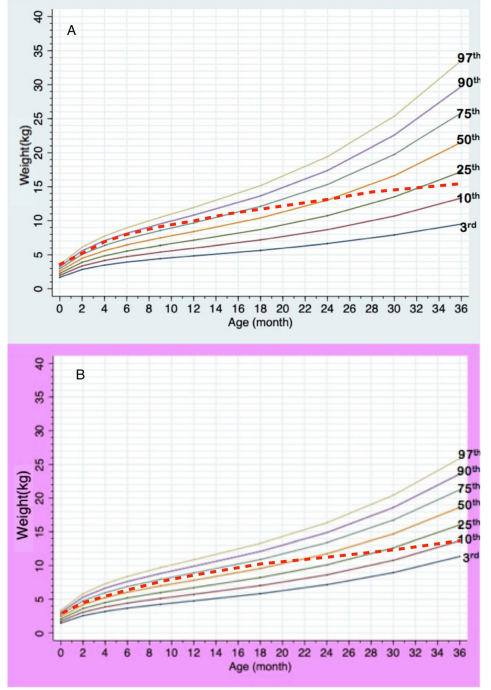
กราฟน้ำหนักตามอายุของเด็กกลุ่มอาการพราเดอร์-วิลลี เพศชาย (A) และเพศหญิง (B)
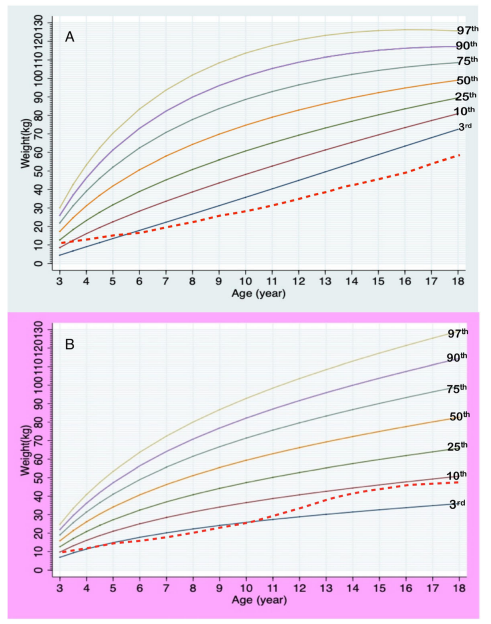
กราฟส่วนสูงตามอายุของเด็กกลุ่มอาการพราเดอร์-วิลลี เพศชาย (A) และเพศหญิง (B)
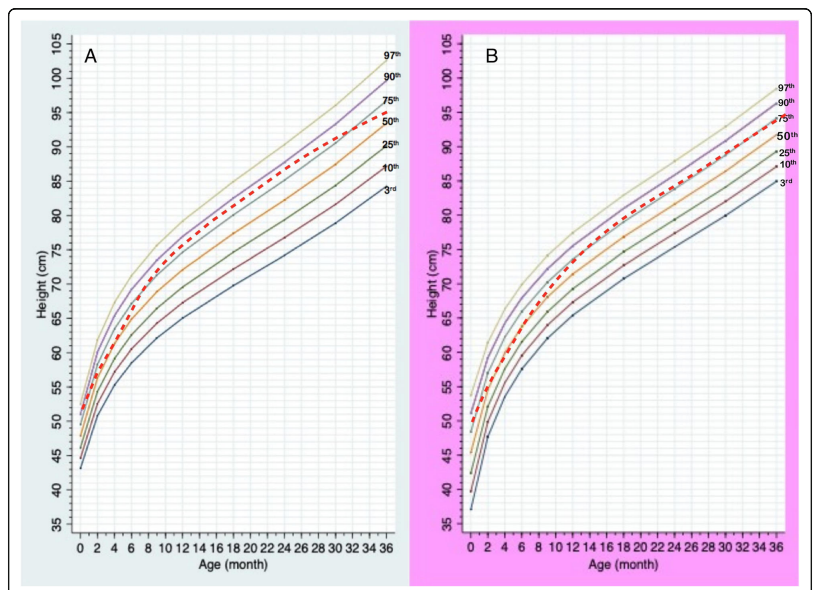
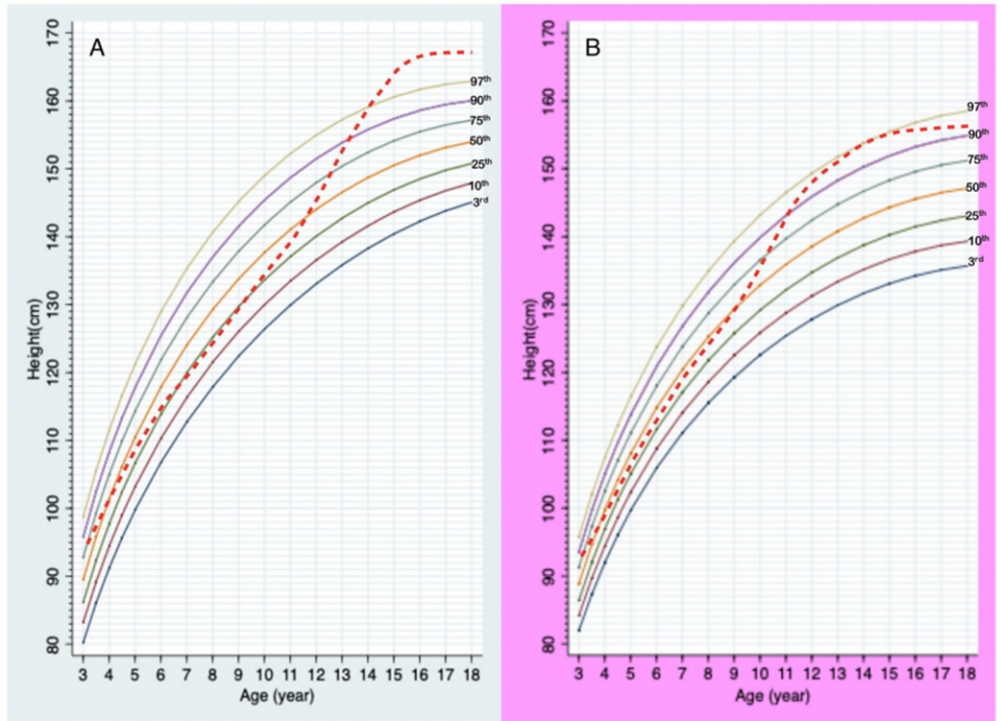
กราฟส่วนสูงตามอายุของเด็กกลุ่มอาการพราเดอร์-วิลลี เพศชาย (A) และเพศหญิง (B)
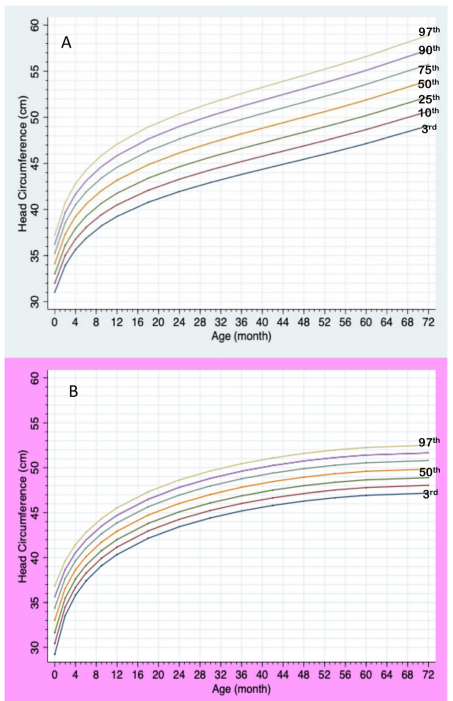
กราฟความยาวรอบศีรษะตามอายุของเด็กกลุ่มอาการพราเดอร์-วิลลี เพศชาย (A) และเพศหญิง (B)
เอกสารอ้างอิง
1. Driscoll DJ, Miller JL, Cassidy SB. Prader-Willi Syndrome. 1998 Oct 6 [Updated 2023 Nov 2]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024. 2. Shawn E. McCandless, The Committee on Genetics; Health Supervision for Children With Prader-Willi Syndrome. Pediatrics January 2011; 127 (1): 195–204. 10.1542/peds.2010-2820 3. Leslie SW, Sajjad H, Villanueva CA. Cryptorchidism. [Updated 2024 May 5]. 4. Jones, Kenneth Lyons. Smith's Recognizable Patterns of Human Malformation. Philadelphia :Saunders, 7th ed. 2013. PP 274-276. 5. Mongkollarp, N., Tim-Aroon, T., Okascharoen, C. et al. Growth charts for Thai children with Prader-Willi syndrome aged 0–18 years. Orphanet J Rare Dis 15, 111 (2020). https://doi.org/10.1186/s13023-020-01388-7
ผู้เขียน นพ. ธรรมกมล อัครธรรม
Update : 19 ส.ค. 2567














